Details for DMR calling
Presegmentation
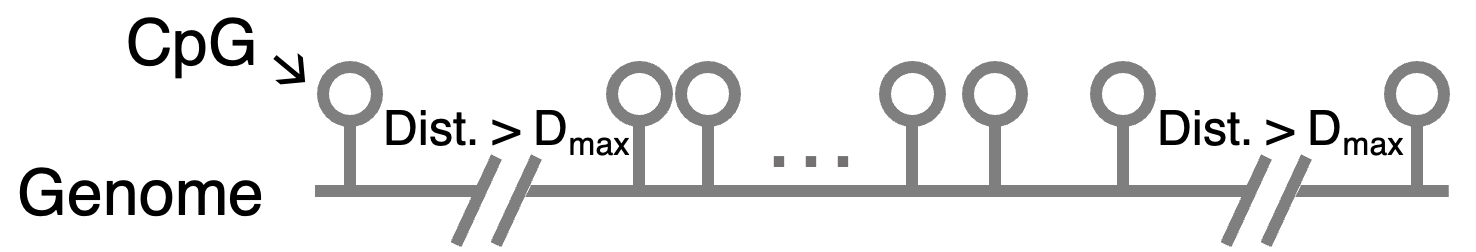
The first step of DMR calling is presegmentation - cutting the genome based on the distances between CpGs. A segment starts with a CpG and stops at a CpG with a distance to the next CpG greater than the max distance allowed \(D_{max}\), which is from -M, --maxdist. Each segment will be further segmented based on methylation data in a thread.
Segmentation
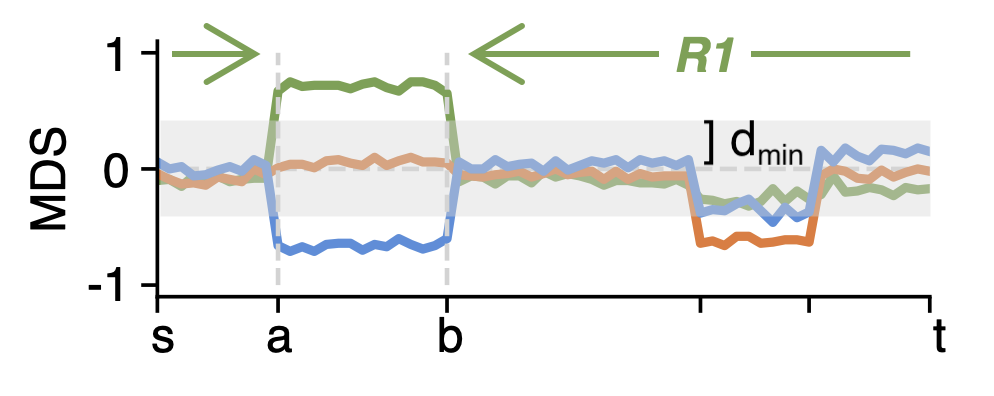
After presegmentation, each segment will be further segmented based on pairwise methylation difference. To be computational effient, filters are applied to skip segments:
- Number of CpGs is lower than \(m_{min}\) from
-m, --minCpGs. - Number of CpGs with absolute mean difference greater than \(d_{min}\) from
-d, --minMethDiff(or-D, --minMethDiffHighwhen calling DMRs without groups) is lower than \(r_{min}\) from-r, --minDMR.
Segments passing the filters will be segmented by finding the \(Z_{max}(s,t,a,b,G,G')\).
Clustering
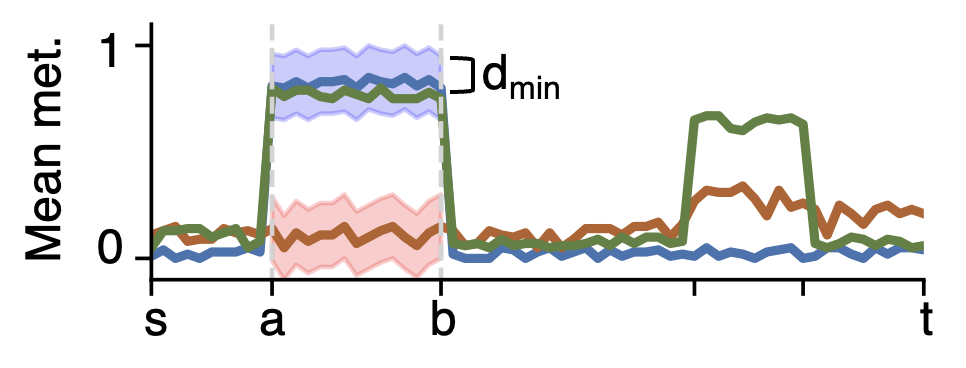
After segmentation, the breakpoint positions \(a,b\) and the pair of groups/samples \(G',H'\) will be used to cluster other groups. A group/sample will be clustered with \(G'\) or \(H'\) according to the methylation-distance. If the group/sample has a ratio of CpGs with minimal difference \(d_{min}\) to \(G' or H'\) smaller than the maximum ratio \(u_{min}\) defined from -u, --clusteringRatio, this group/sample will be clustered with \(G' or H'\). Otherwise, this group/sample will be marked as intermediate. If the clustering is unambiguous, this group/sample will also be marked as intermediate.
Recursion
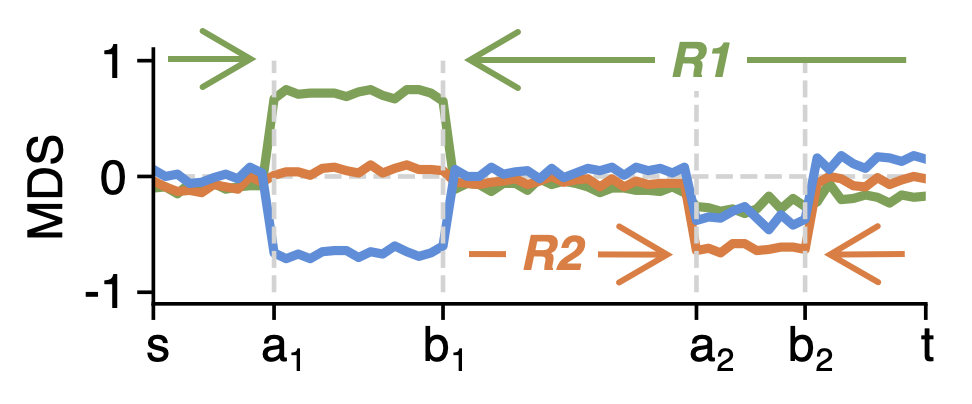
After the first breakpoint pair \(a,b\) is found, metilene3 will continue to further segment the sub-regions \([s,a_1]\), \([a_1,b_1]\) and \([b_1,t]\), until the criteria of termination are met:
- Number of CpGs in a sub-region is lower than \(m_{min}\) from
-m, --minCpGs. - No p-values of all sub-regions is found more significant than the p-value of the region to be further segmented.